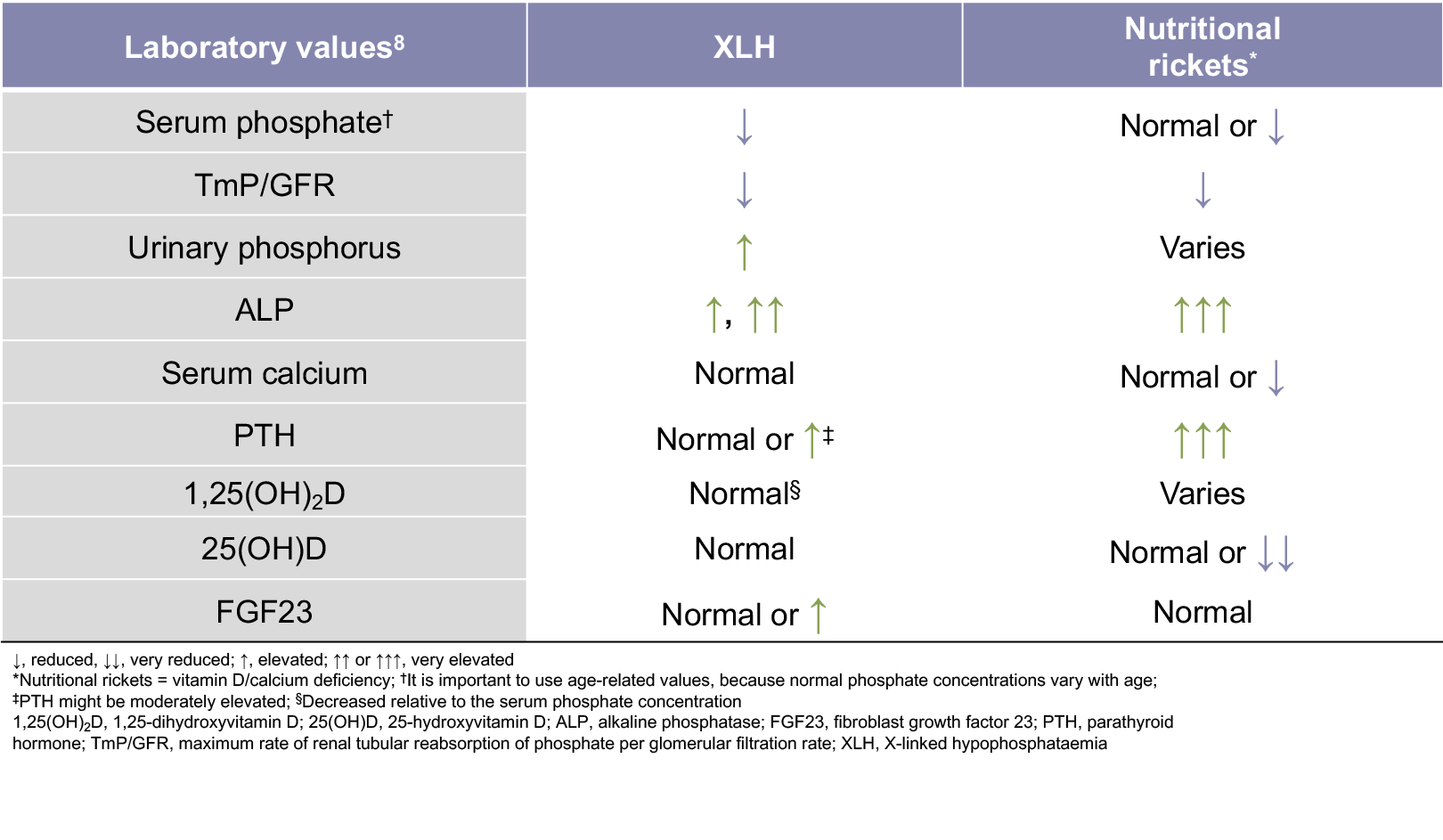
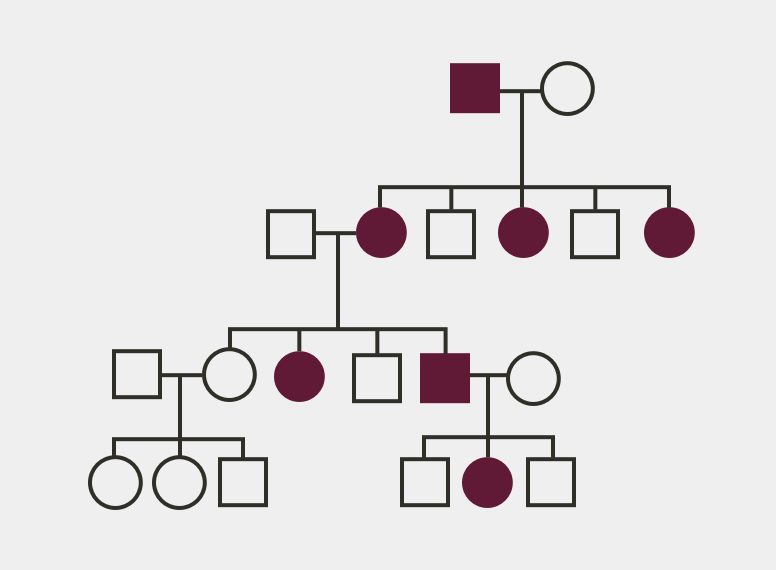
1. Ruppe MD. X-linked hypophosphatemia. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. Gene Reviews. https://www.ncbi.nlm.nih.gov/books/NBK83985/. Accessed October 20, 2017. 2. Econs MJ, Samsa GP, Monger M, Drezner MK, Feussner JR. X-linked hypophosphatemic rickets: a disease often unknown to affected patients. Bone Miner. 1994;24(1):17-24. 3. Linglart A, Biosse-Duplan M, Briot K, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect. 2014;3(1):R13-R30. 4. Skrinar A, Dvorak-Ewell M, Evins A, et al. The lifelong impact of X-linked hypophosphataemia: results from a burden of disease survey. J Endocr Soc. 2019;3(7):1321-1334. 5. Hardy DC, Murphy WA, Siegel BA, Reid IR, Whyte MP. X-linked hypophosphatemia in adults: prevalence of skeletal radiographic and scintigraphic features. Radiology. 1989;171(2):403-414. 6. Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician’s guide to X-linked hypophosphatemia. J Bone Miner Res. 2011;26(7):1381-1388. 7. Santos F, Fuente R, Mejia N, Mantecon L, Gil-Peña H, Ordoñez FA. Hypophosphatemia and growth. Pediatr Nephrol. 2013;28(4):595-603. 8. Haffner D, Emma F, Eastwood DM, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019;15(7):435-455. 9. Payne RB. Renal tubular reabsorption of phosphate (TmP/GFR): indications and interpretation. Ann Clin Biochem. 1998;35(pt. 2):201-206. 10. Goldsweig BK, Carpenter TO. Hypophosphatemic rickets: lessons from disrupted FGF23 control of phosphorus homeostasis. Curr Osteoporos Rep. 2015;13(2):88-97. 11. Imel EA, Carpenter TO. A practical clinical approach to paediatric phosphate disorders. Endocr Dev. 2015;28:134-161. 12. Özkan B. Nutritional rickets. J Clin Res Pediatr Endocrinol. 2010;2(4):137-143. 13. Nield LS, Mahajan P, Joshi A, Kamat D. Rickets: not a disease of the past. Am Fam Physician. 2006;74(4):619-626. 14. Beck-Nielsen SS, Brusgaard K, Rasmussen LM, et al. Phenotype presentation of hypophosphatemic rickets in adults. Calcif Tissue Int. 2010;87(2):108-119. 15. Beck-Nielsen SS, Brixen K, Gram J, Brusgaard K. Mutational analysis of PHEX, FGF23, DMP1, SLC34A3 and CLCN5 in patients with hypophosphatemic rickets. J Hum Genet. 2012;57(7):453-458. 16. Gaucher C, Walrant-Debray O, Nguyen T-M, Esterle L, Garabédian M, Jehan F. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum Genet. 2009;125(4):401-411. 17. Whyte MP, Schranck FW, Armamento-Villareal R. X-linked hypophosphatemia: a search for gender, race, anticipation, or parent of origin effects on disease expression in children. J Clin Endocrinol Metab. 1996;81(11):4075-4080.