Diagnosis
A diagnosis of XLH is typically based on clinical and biochemical findings in combination with family history; however, variations in disease presentation can lead to delayed diagnosis or misdiagnosis.1,2
Molecular genetics can be used to establish a diagnosis, determine if XLH is inherited, and what risk there is to family members.1
Clinical
In children:
Children with XLH typically present with lower-extremity bowing or genu valgum, impaired growth, and gait abnormalities during the first 1 to 2 years of life. However, diagnosis may occur after the age of 2 or even in adulthood.1,3
In adults:
Biochemical
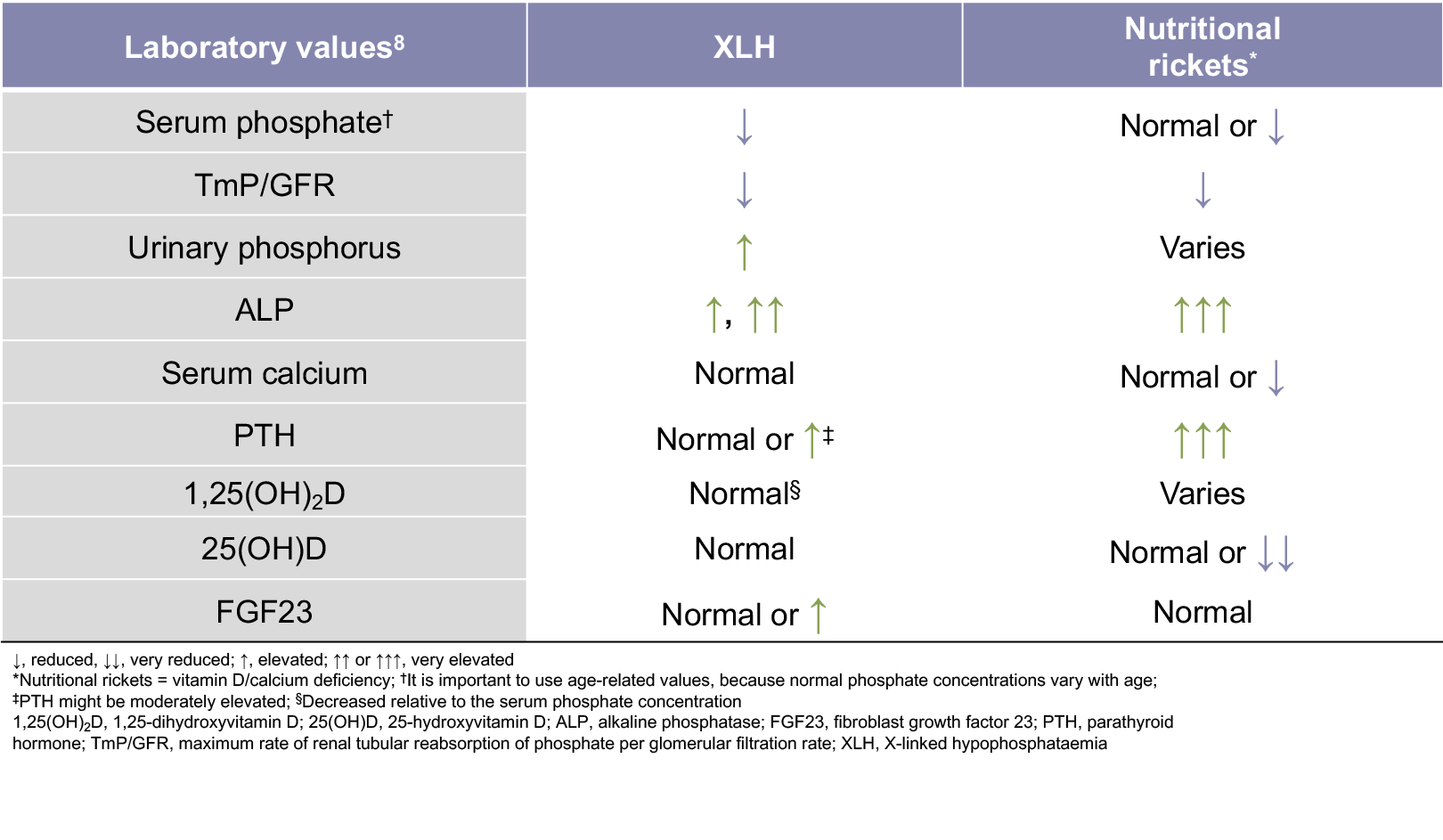
The main biochemical features of XLH are low serum phosphate levels, decreased 1,25-dihydroxyvitamin D levels relative to the serum phosphate concentration, a reduced ratio of tubular maximum reabsorption of phosphate to glomerular filtration rate (TmP/GFR), and elevated serum FGF23 levels.1,6,8
- Additional biochemical features of XLH include normal 25-hydroxyvitamin D levels, elevated urinary phosphorus levels, elevated alkaline phosphatase levels, and elevated or normal parathyroid hormone levels1,7,8
The TmP/GFR is the ratio of renal tubular maximum reabsorption of phosphate (TmP) to glomerular filtration rate (GFR)
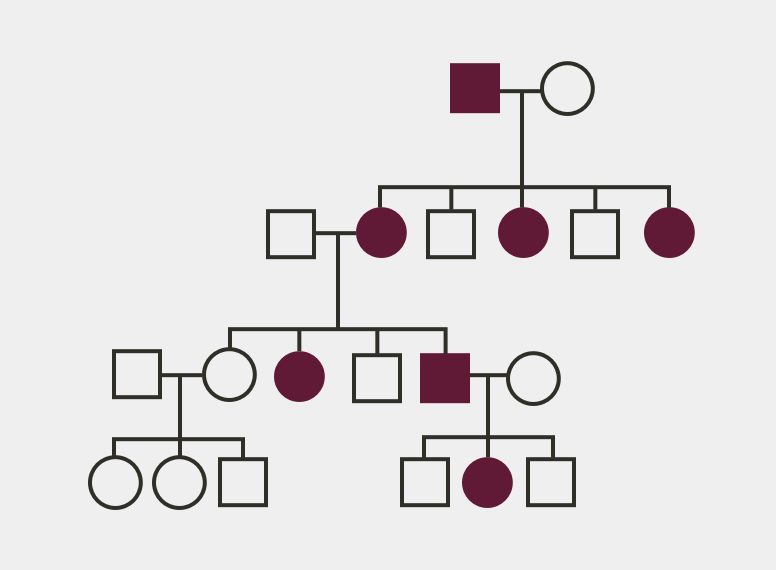
Family History
- Evaluation of at-risk infants and children is warranted to ensure early diagnosis and treatment, which has been shown to improve clinical outcomes3
- Screening of family members of XLH patients may help to identify previously undiagnosed individuals14
Pedigree Analysis